The MHRA states that the new roadmap will ensure UK patients have timely access to technology while also securing the UK’s position as a world leader for medtech innovators.
Dr Laura Squire, the MHRA’s MedTech Regulatory Reform Lead and Chief Officer Healthcare, Quality and Access states: “The roadmap sets out how we will work with stakeholders including patients as the process moves forward, giving early sight of what is to come and giving us feedback about the guidance they will need, to ensure the successful implementation of these wide-ranging UK reforms.”
It is expected that the planned regulations will also help the UK align with international partners, as a result of more patient-minded and proportionate requirements for medical devices that are responsive to technological advances.
The future core regulations will introduce numerous requirements that will impact on the current systems in place for manufacturers serving the Great Britain market. Many of these planned core regulations are similar to those seen with the current updated European Device Regulations. Of note is the plan to bring the essential requirements for medical devices being placed on the market in Great Britian into a greater alignment with those of the EU, including cybersecurity requirements for software as a medical device, which includes for artificial intelligence. For manufacturers, developers and patients, this should be a welcome approach. Similar to EU regulation will be the requirement to have a Person Qualified in Regulatory Compliance and a clarification around requirements for economic operators; manufacturers, importers and distributors.
There will be a strengthening of the requirements for quality management systems and technical documentation including the introduction of a need for devices to have a unique device identifier (UDI). Other steps to be brought in are new requirements for clinical investigations.
For implantable devices, the core regulations will see an up-classifying leading to more stringent pre and post market requirements with the requirement to provide implant cards (enabling patients to know which device they have had implanted).
In addition, a change to the classification of several types of devices, with focus on increasing the class of certain software as a medical device with an alignment of IVD classifications with those of the International Medical Device Regulators Forum.
Other introductions are to clarify the requirements for conformity assessments and approved bodies, introduce a framework for international recognition, enabling swifter access for devices already approved by comparable regulators as well as for those who have Medical Device Single Audit Program (MDSAP) certificates. Furthermore, new requirements for exempt in-house manufactured devices and custom-made devices will be introduced. There will be new requirements around the claims manufacturers can make about their medical devices requiring them to align with their statement of intended purpose.
In addition, in 2024 the UK government plan to introduce legislation to strengthen Post-Market Surveillance requirements (PMS) in GB ahead of the wider future regulatory regime.
A Draft PMS Statutory Instrument (SI) was made available for consultation in July 2023 with the World Trade Organisation (WTO) published notification and included:
- Detail on what must be included as part of a PMS system, including the methods for collecting PMS data to support improved capturing of PMS data and harmonisation across manufacturers.
- Enhanced serious incident reporting obligations for manufacturers to support the detection of safety issues sooner.
- More stringent requirements for manufacturers to conduct periodic reviews of their PMS data, including for implantable medical devices. This aims to support manufacturers in earlier detection of trends/signals that may have an impact on the safety of a medical device.
The consultation period closed in September 2023 and the final PMS SI is to be laid in the first part of 2024 and expected to apply towards the end of 2024.
Timings
To outline the planned implementation of medical devices future regime, the MHRA has published a roadmap presenting the intended timelines for delivering the future regulatory framework. During the first 6 months of 2024, MHRA plan to carry out stakeholder discussions with regard to the future core regulations developed from the policy positions in the previous regulatory device consultation response. Improvements to the regulatory framework for medical devices will be made over the next two years, with “priority measures” to patient safety set for this year.
The regulations will be delivered through four SIs. It is intended that priority measures to enhance PMS will be put in place first in 2024, with core elements of the new framework expected to be in place in 2025.
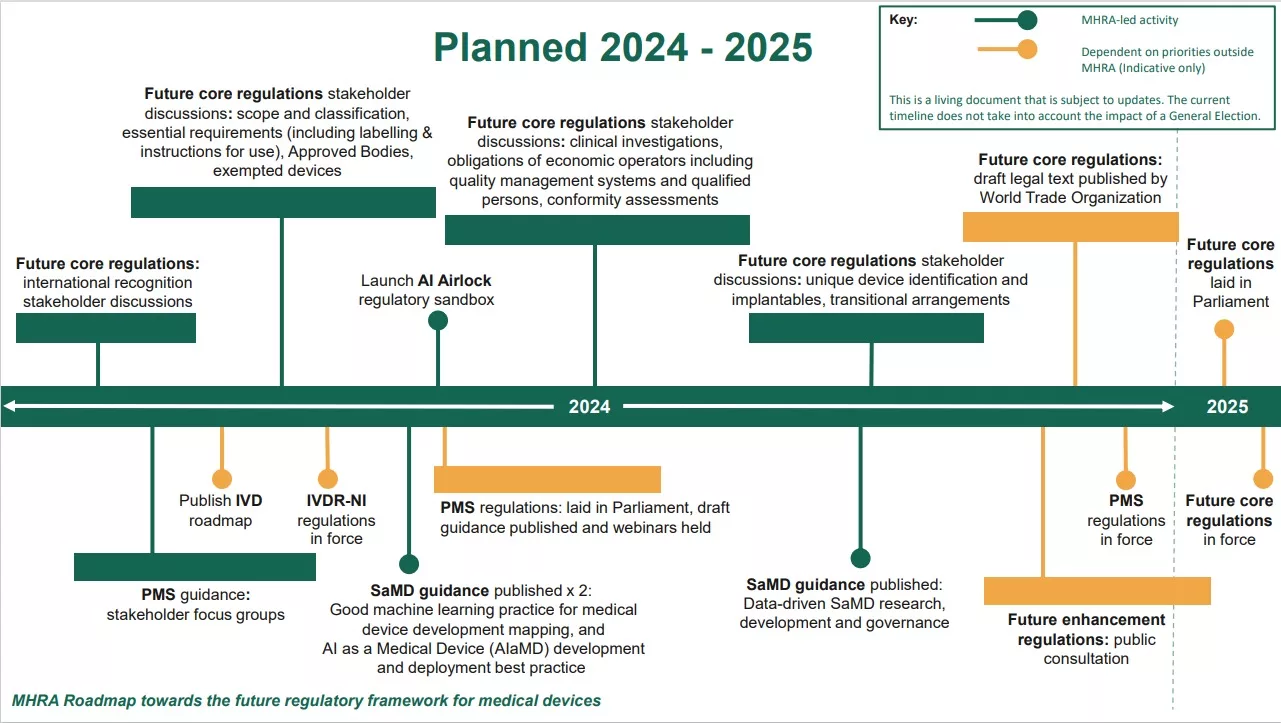
Source: MHRA Roadmap towards the future regulatory framework for medical devices
The MHRA states that these future core regulations will enable greater international collaboration and practices, with more patient-centred, proportionate requirements for medical devices which are responsive to technological advances. The Agency is working with industry trade associations and the wider medical devices community to support the effective implementation of the new regulations and guidance will be issued to support the interpretation of the new regulatory requirements.
Considerations for Medical Device Manufacturers
Overall, the changes should make it easier for organisations to navigate the regulatory pathway and help to clear up issues relating to misalignment with the EU’s requirements. It will be key for businesses to plan out their regulatory strategy early in the development process, particularly for those devices with a combinational aspect, or involving software and AI. UK medical device manufacturers should ensure their quality management systems, device development processes, and post-market surveillance are appropriate and in line with any new regulations and should be cognisant of upcoming requirements. Medical device regulation is a complex area, and seeking the support of an expert is advisable to ensure compliance as the UK moves towards harmonisation and alignment of device regulation.
If you have any questions or would like to discuss the “Roadmap towards the future regulatory framework for medical devices” further, get in touch with one of our regulatory medical device experts today.







